Спадкові захворювання нервової системи, м'язові дистрофії, міастенія. Захворювання кістякових м'язів.
Нервово-м'язові захворювання (НМЗ) – це найбільш численна група спадкових захворювань, в основі яких лежить генетично детерміноване ураження передніх рогів спинного мозку, периферичних нервів та скелетних м'язів.
До нервово-м'язових захворювань відносяться:
1) прогресуючі м'язові дистрофії (первинні міопатії);
2) спинальні та невральні аміотрофії (вторинні міопатії);
3) уроджені непрогресуючі міопатії;
4) нервово-м'язові захворювання з міотонічним синдромом;
5) пароксизмальні міоплегії;
6) міастенія.
15.2. Прогресують м'язові дистрофії (первинні міопатії)
Прогресують м'язові дистрофії (ПМД),або первинні міопатії, що характеризуються дегенеративними змінами в м'язовій тканині.
Патоморфологічні змінипри ПМД характеризуються витонченням м'язів, заміною їх жировою та сполучною тканиною. У саркоплазмі виявляються осередки фокального некрозу, ядра м'язових волокон розташовуються ланцюжками, м'язові волокна втрачають поперечну смугастість.
Питання патогенезу залишаються дотепер невирішеними. В основі міопатії лежить дефект мембрани м'язових клітин. Великі сподівання покладаються на молекулярну генетику.
Різні форми міопатії відрізняються типом успадкування, термінами початку процесу, характером та швидкістю його перебігу та топографією м'язових атрофій.
Міопатії клінічно характеризуються слабкістю та атрофією м'язів. Існують різні форми ПМД.
15.2.1. Міодистрофія Дюшенна (псевдогіпертрофічна форма пмд)
Зустрічається найчастіше із усіх ПМД (30:100 000). Дана форма характеризується раннім початком (2-5 років) та злоякісною течією, хворіють переважно хлопчики. Міопатія Дюшенна успадковується за рецесивним типом, зчепленим з Х-хромосомою. Патологічний ген локалізується у короткому плечі хромосоми (X, або 21 хромосоми).
Досить висока мутація гена, чим пояснюється значна частота спорадичних випадків. Мутація (найчастіше делеція) гена призводить до відсутності дистрофіну в мембрані м'язових клітин, що призводить до структурних змін сарколеми. Це сприяє виходу кальцію і веде до загибелі міофібрилу.
Однією з перших ознак захворювання є ущільнення литкових м'язів та поступове збільшення їх обсягу за рахунок псевдогіпертрофій. Процес носить висхідний характер. Для розгорнутої стадії захворювання характерна «качина» хода, хворий ходить, перевалюючись з боку на бік, що пов'язано головним чином зі слабкістю м'язів сідниць.
Внаслідок цього відбувається нахил таза у бік неопорної ноги (феномен Тренделенбурга) і компенсаторний нахил тулуба у протилежний бік (феномен Дюшенна). При ходьбі сторона нахилу постійно змінюється. Це можна перевірити в позі Тренделенбурга, попросивши хворого підняти одну ногу, зігнувши її під прямим кутом у колінному та кульшовому суглобі: таз на боці піднятої ноги опускається (а не піднімається як у нормі) через слабкість середнього сідничного м'яза опорної ноги.
При міопатії Дюшенна часто відзначається виражений лордоз, крилоподібні лопатки, типові м'язові контрактури, рано випадають колінні рефлекси. Нерідко вдається виявити зміни у кістковій системі (деформацію стоп, грудної клітки, хребта, дифузний остеопороз). Може відзначатись зниження інтелекту та різні ендокринні розлади (адипозогенітальний синдром, синдром Іценка-Кушинга). До 14-15 років хворі зазвичай вже повністю знерухомлені, у термінальній стадії слабкість може поширитися на м'язи обличчя, горлянки, діафрагми. Гинуть вони найчастіше на 3-му десятилітті життя від кардіоміопатії або приєднання інтеркурентних інфекцій.
Відмінною рисою міопатії Дюшенна є різке підвищення специфічного м'язового ферменту - креатинфосфокінази (КФК) у десятки та сотні разів, а також підвищення міоглобіну у 6-8 разів.
Для медико-генетичного консультування важливим є встановлення гетерозиготного носійства. У 70% гетерозигот визначаються субклінічні та клінічні ознаки м'язової патології: ущільнення та збільшення литкових м'язів, швидка стомлюваність м'язів при фізичному навантаженні, зміна м'язових біоптатів та біопотенціалів за даними ЕМГ.
Сторінка 44 з 44
Скелетні м'язи залучаються до патологічного процесу при різноманітних дегенеративних, обмінних та запальних захворюваннях. Найчастіше у своїй відбувається дегенерація м'язових волокон, а за хронічних формах - їх заміщення сполучною тканиною і жиром. Проксимальні групи м'язів пошкоджуються значно, ніж дистальні, як і нижні кінцівки по відношенню до верхніх. Хвора дитина відрізняється так званою качиною (ходом, що перевалюється), не здатна бігати, підніматися сходами і вставати, якщо знаходиться в положенні сидячи. Сухожильні рефлекси у нього пригнічені, ступінь їх згасання пропорційний до ступеня ослаблення м'язової сили. Чутливість не порушується.
До діагностично цінних лабораторних методів належить визначення активності ферментів, особливо креатинфосфокінази, у сироватці. Цей фермент, що каталізує реакцію: фосфокреатин + АДФ-креатин + АТФ, присутній головним чином у клітинах головного мозку та м'язової тканини. При деяких дифузних м'язових захворюваннях, особливо при м'язовій дистрофії, його надлишкові кількості проникають у міжклітинний простір та кров. У хворих зазвичай підвищена активність сироваткової лактатдегідрогенази та глутамінощавелевооцтової трансамінази, проте широкий розподіл їх в інших тканинах, включаючи печінкову, зменшує специфічність тесту. Зазвичай для уточнення діагнозу потрібна біопсія м'язової тканини.
Запальні хворобим'язів. Запалення м'язової тканини супроводжує деякі інфекції, особливо трихінельоз, токсоплазмоз та спричинені вірусом Коксакі. Нерідко воно буває компонентом колагенових хвороб, у тому числі дерматоміозиту, червоного вовчака, вузликового періартеріїту та ревматоїдного артриту.
Поліміозит. Дифузне ізольоване запалення м'язів невідомої етіології називають поліміозит. Для нього типові швидкий прогресуючий перебіг, слабкість та біль у проксимальних групах м'язів. Часто в процес залучаються м'язи шиї, у зв'язку з чим дитині важко підняти голову і утримувати її в цьому положенні. До лабораторних ознак запалення м'язів відноситься збільшення ШОЕ та числа лейкоцитів. Проте їхня відсутність не виключає поліміозиту. Рівень сироваткових ферментів зазвичай підвищений. У м'язовому біоптаті визначають дегенерацію та часткову регенерацію волокон та їх інфільтрацію лімфоїдними клітинами. Диференціювати поліміозит від м'язової дистрофії та дерматоміозиту важко. Він може являти собою атипову формудерматоміозиту, хоча гістологічна картина при цих двох станах дещо різна: для дерматоміозиту характерний васкуліт, який зазвичай відсутній при поліміозиті. Прогноз при останньому дещо сприятливіший. Лікування кортикостероїдами супроводжується ефектом, але при їх скасуванні може настати рецидив.
Прогресуючий оссифицирующий міозит. Етіологія цього захворювання, що рідко зустрічається. сполучної тканинита м'язів невідома. Повідомляється, що на них страждають рідні брати та сестри, у тому числі близнюки, і передається воно кровним родичам по прямій лінії. Припускають, що успадковується хвороба за аутосомно-домінантним типом. Хлопчики хворіють у 2-3 рази частіше за дівчаток.
Патологічні ознаки залежить від стадії захворювання. На ранніх стадіях місцеві набряки та запальні клітинні інфільтрати знаходять у м'язах та сухожиллях. Пізніше ділянки запалення заміщаються грануляційною тканиною і в кінці вогнищах ураження формуються ділянки хрящової та кісткової тканини.
Майже у 75% хворих дітей виявляють уроджені вади розвитку, найчастіше недорозвинення пальців та анкілоз фаланг I пальців ніг та недорозвинення I пальців рук, полідактилію, викривлення пальців, синдактилію (ноги), деформацію вушних раковин, глухоту, відсутність зубів Ті ж уроджені вади можуть бути у родичів хворого, у яких не розвинулося прогресуюче захворювання сполучної тканини та м'язів. Вік, у якому може початися асифікуючий міозит, варіює від народження до старшого дитячого віку. Зазвичай розрізняють три стадії його: 1) на місцях незначних місцевих травм з'являються обмежені, часто теплі та м'які на дотик тістоподібні припухлості м'яких тканин; 2) через кілька днів симптоми запалення зникають, а вогнище ураження твердне; 3) відбувається окостеніння ураженої ділянки. Періодично з'являються нові осередки, переважно в областях шиї та спини. Первинною ознакоюможе стати кривошою, якщо процес розвинувся в грудиноключично-соскоподібному м'язі. Зрештою окостеніння поширюється на багато сухожилля та зв'язки. Настає анкілоз хребта та суглобів рук та ніг (рис. 21-5). Запалення може поширитися на скронево-нижньощелепні суглоби, у зв'язку з чим утруднюються жувальні рухи. Кісткові вирости можуть виступати через шкіру. У юнацькому віці захворювання часто призводить до повного знерухомлення та смерті внаслідок дихальної недостатності та припинення дихання, хоча є повідомлення про випадки виживання. При оссифицирующем міозиті велика небезпека розвитку остеогенної саркоми.
Рис. 21-5. Дитина з прогресуючим асифікуючим міозитом (типова поза з ригідністю м'язів шиї та спини).
Іноді патологічний процес буває обмежений місцем травми м'яких тканин, що передувала (miositis ossificans circumscripta). Широко поширена кальцифікація м'язової тканини може відбутися і при хронічному поліміозіті та дерматоміозіті.
Результати лабораторних методівдосліджень немає діагностичної цінності.
Рівні у сироватці кальцію, фосфору, лужної фосфатази, а також активність креатинфосфокінази та інших ферментів залишаються в нормі. Кісткова тканинау вогнищі ушкодження не відрізняється за будовою від норми.
Існуючі методи лікування є незадовільними. У деяких випадках відмічено уповільнення розвитку захворювання при застосуванні АКТГ та інших кортикостероїдів. Їхня роль у кінцевому результаті лікування викликає сумніви.
Ендокринні та обмінні міопатії. Міопатія при гіпертиреозі є досить рідкісним ускладненням. Для нього типові птоз, двосторонній парез лицьових м'язів та м'язів проксимальних відділів кінцівок. При цьому деякі симптоми гіпертиреозу можуть маскуватися м'язовою слабкістю, однак залишаються тахікардія, посилене потовиділення і збільшення щитовидної залози. Сухожильні рефлекси на відміну багатьох інших форм міопатії залишаються у нормі. Після корекції гіпертиреозу м'язова слабкість поступово зникає.
Міопатія при гіпотиреозі. Гіпотиреоз у немовлят може бути пов'язаний з м'язовими слабкістю та гіпотонією. У дітей старшого дитячого віку з мікседемою сповільнюються скорочення та розслаблення м'язів, у деяких випадках відзначається м'язова гіпертрофія (синдром Дебре – Семелена). Сукупність таких ознак, як слабкість та гіпертрофія м'язів, дає можливість припустити м'язову дистрофію.
Міопатія при лікуванні кортикостероїдами. Вона може ускладнювати хворобу Іценка – Кушинга, але частіше розвивається при лікуванні більшими дозами синтетичних стероїдів. Слабкість особливо помітна в м'язах тазового пояса, що проявляється в ході, що перевалюється (качині), утрудненнях при підйомі по сходах і спробі встати з положення сидячи. Колінний рефлекс відсутній. Може настати стоншення м'язів. Міопатичні зміни в м'язовій тканині зазвичай незначні навіть за її вираженої слабкості. М'язова сила після відміни кортикостероїдів відновлюється повільно (протягом кількох місяців).
Міопатія при гіперпаратиреозі. Гіперпаратиреоз може бути пов'язаний зі слабкістю та гіпорефлексією, зумовленими гіперкаліємією. Зазвичай вони швидко зникають після паратиреоїдектомії.
Дефіцит карнітину (ліпідна міопатія) супроводжує накопичення у великих кількостях ліпідів у м'язах та порушення у зв'язку з цим енергетичного забезпечення останніх. Карнітин відноситься до обов'язкових компонентів системи, що забезпечує перенесення жирних кислот з довгим ланцюгом з цитозолю в мітохондрії, де вони окислюються. М'язова слабкість розвивається за двох форм недостатності карнітину.
Недостатність карнітину в м'язах клінічно представлена прогресуючою слабкістю їх проксимальних груп, частіше у школярів та підлітків. Іноді слабкість інтермітує та поєднується з міоглобінурією. При тяжкому перебігу може настати параліч дихальної мускулатури. Рівень ферментів (креатинкіназу та альдолазу) у сироватці підвищується. На електроміограмі виявляють неспецифічні зміни, властиві міопатії. У біоптаті м'язів можна побачити велику кількість крапель жиру. Рівень карнітину в сироватці не змінюється, але у м'язах знижується. Розпізнавання патології має важливе значення, оскільки вона може бути курабельною. Нерідко її сприймають як м'язову дистрофію. Ефект може настати після застосування внутрішньо 100 мг/(кг/добу) карнітину. У деяких випадках ефективне лікування кортикостероїдами.
- Системна недостатність карнітину проявляється прогресуючою міопатією, у тому числі кардіоміопатією, та дисфункцією печінки, що супроводжується клінікою печінкової енцефалопатії на кшталт синдрому Рея. Від останнього карнітінова недостатність відрізняється рецидивуючим перебігом і енцефалопатії, що зберігається між періодами загострення, вираженою м'язовою слабкістю. Рівень креатинфосфокінази у сироватці помітно підвищений, кількість карнітину зменшено як у сироватці, так і у м'язах. Зміни в біоптаті аналогічні таким при дефіцит карнітину в м'язовій тканині. Подібні клінічні та морфологічні зміни, у тому числі карнітинову недостатність, можна виявити при порушенні обміну органічних кислот, наприклад, при метилмалонової та глутарової ацидурії (вторинна карнітинова недостатність).
Рис. 21-6. Дитина з вродженою відсутністю лівого великого грудного м'яза. 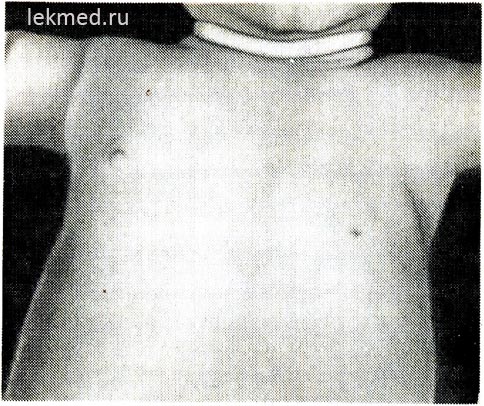
Привертають увагу відсутність передньої пахвової складки і низкорасположенный сосок.
Лікування полягає у дотриманні хворим на дієту, багату на вуглеводи і знежирену, і прийом карнітину в добової дози 100 мг/кг.
Уроджені м'язові дефекти. Вроджена відсутність м'яза. Недорозвинення м'язів може бути досить поширеним і спричиняти повну блокаду рухів у суглобах або вродженому артрогрипозу. Як уроджений дефект найчастіше відсутній один м'яз. До досить поширеної аномалії відноситься відсутність стернальної частини великого грудного м'яза (рис. 21-6), у деяких випадках цей дефект поєднується з синдактилією на ураженому боці (синдром Поланда). Відсутність грудного м'яза часто супроводжує дистрофію м'язів. Вроджена відсутність черевних м'язів живота часто пов'язана із дефектами розвитку сечових шляхів.
Рис. 21-7. Деформація шиї та асиметрія обличчя у хлопчика з вродженою кривошиєю, не лікованого з 12-річного віку. 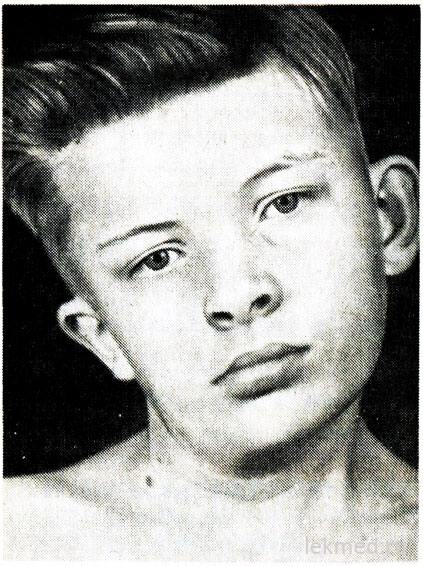
Вроджена кривошия обумовлена одностороннім укороченням або контрактурою грудиноключично-соскоподібного м'яза. Голова хворого нахилена у бік контрактури, а підборіддя спрямоване донизу у протилежний бік (рис. 21-7). При спробі корекції положення голови відчувається значний опір м'язів. У ураженому м'язі промацуються ділянки ущільнення. Причина дефекту незрозуміла, протягом тривалого часу його вважали результатом пологової травми. Однак кривошия зустрічається у дітей, народжених за допомогою операції кесаревого розтину; це дає можливість припустити, що в деяких випадках причина дефекту відноситься до внутрішньоутробного періоду. Кривошею слід диференціювати від патологічного нахилу голови внаслідок деформації шийних хребців, наприклад при аномалії Кліппеля – Вейля, та від переломів чи вивихів шийних хребців. Їх виключають з допомогою рентгенографічного дослідження. У дітей старшого віку нахил голови може бути при косоокості, дистонії, пухлинах задньої черепної ямки та шийного відділу спинного мозку, що осифікує міозит, шийному лімфаденітіабо діафрагмальної грижі. У більшості випадків вроджена кривошия піддається корекції за допомогою лікувальної гімнастики. Однак при хронічній формікривошия призводить до асиметричного розвитку обличчя та голови (див. рис. 21-7), у зв'язку з чим може знадобитися розтин м'язу, що викликано косметичними цілями.
Вроджені міопатії. У цю групу входять кілька рідкісних форм успадкованих захворювань, при яких м'язові слабкість та гіпотонія з'являються з грудного віку (див. табл. 22-1). Їх точна діагностика має велике значенняз погляду прогнозу. Загалом для нормальної життєдіяльності та тривалості життя він сприятливий на відміну від хвороби Вердніга – Гоффманна чи вродженої м'язової дистрофії. Виявленню вроджених міопатій зазвичай сприяє біопсія м'язів.
- Хвороба центрального стрижня. Центральна частина м'язових волокон забарвлена аномально, але однорідно. При електронномікроскопічному дослідженні виявляють зменшення кількості мітохондрій та збіднення саркоплазматичного ретикулуму у центральній частині волокон.
Немалінова міопатія. Термін «немалінова» пояснюється тим, що в м'язових волокнах визначаються ниткоподібні структури.
Рис. 21-8. Міотонічне скорочення язика (а) при різкому ударі перкусійним молоточком по його правій половині та вік (б) у дитини з гіперкаліємічною формою сімейного періодичного паралічу. 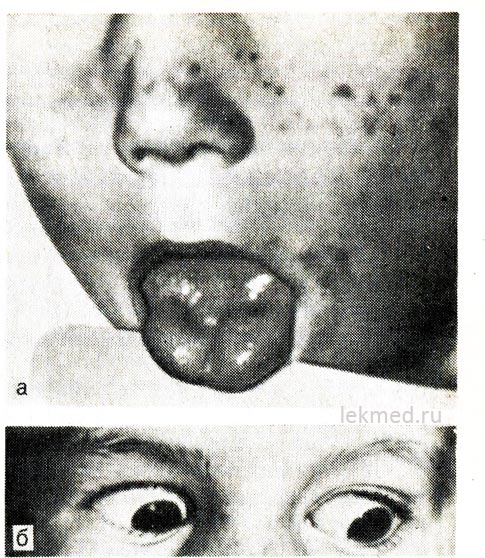
При погляді вниз повік залишається скороченим.
Дані електронно-мікроскопічного дослідження свідчать, що це результат змін Z-смужок міофібрил.
Мітохондріальні міопатії. Повідомляється про деякі форми міопатій, при яких найбільше важливі змінивідбуваються в мітохондріях м'язових волокон. Вони можуть помітно збільшуватись як у числі, так і в розмірах. М'язові слабкість та гіпотонія можуть визначатися вже у грудному віці, але іноді помітно прогресують лише у шкільному. Кардіоміопатія, енцефалопатія та лактатацидемія часто супроводжують міопатії цієї групи.
Міотонія. Цей стан є ознакою різних м'язових захворювань, наприклад дистрофічної міотонії, гіперкаліємічної форми сімейного пароксизмального паралічу та хвороб накопичення глікогену. Міотонія визначається як значне уповільнення розслаблення м'язів після їх довільних чи вимушених скорочень. Клінічно вона проявляється в нездатності розтиснути кулак або у видимому тривалому скороченні м'язів слідом за їх стимуляцією, що виражається в різкому подразненні (рис. 21-8). Це можна спостерігати, якщо вдарити перкусійним молоточком по поверхневій групі м'язів, наприклад, по м'язах язика або долонної поверхні в області підвищення I пальця. Міотонія підтверджується даними електроміографії. У цьому випадку помітна характерна спонтанна активність м'язів після їхнього розслаблення або довільного скорочення (міотонічні розряди).
Вроджена міотонія (хвороба Томсена). Єдиною ознакою цього захворювання, що успадковується за домінантним типом, є міотонія. Воно може виявитися в грудному віці у вигляді уповільнення ковтальних рухів і блювання услід-
ня нездатності до нормального розслаблення м'язів глотки. У старшому дитячому віціМіотонія проявляється як нездатність хворого розтиснути стислі в кулак пальці. За першої спроби здійснити якийсь рух м'язи дитини стають твердими. При багаторазовому повторенні цього руху вони дещо розслабляються. Так, наприклад, хвора дитина зазнає великих труднощів на початку акту ходьби. Перші кілька кроків він зазвичай здійснює дуже невпевнено та повільно. Через кілька секунд хода стає нормальною або майже нормальною. Симптоми міотонії посилюються при несприятливому емоційному стані хворого та охолодженні організму. М'язова сила залишається в нормі, м'язи досить розвинені і нерідко помітно збільшені, що створює хибне враження атлетичного складання хворого.
Діагноз ґрунтується на клінічних даних та даних електроміографії. Активність сироваткових ферментів перебуває у межах норми. Єдиною гістологічною ознакою є гіпертрофія м'язових волокон.
Від дистрофічної міотонії захворювання відрізняється відсутністю м'язових слабкостей та атрофії та дистрофічних змін у біоптаті м'язової тканини. Лікування новокаїном або сульфатом хінідину супроводжується ефектом та показано при функціональних порушеннях. Перебіг захворювання зазвичай доброякісний, і стан хворого може з віком покращуватись.
Пароксизмальні паралічі.Для цієї групи захворювань характерна періодична м'язова слабкість із повним чи майже повним відновленням сили м'язів у період між нападами. До неї відноситься і дефіцит м'язової фосфорилази (хвороба Макардла).
Гіперкаліємічний пароксизмальний параліч.Спадкова епізодична адинамія, або параміотонія, передається за домінантним типом і особливо важко протікає в осіб чоловічої статі. Починається зазвичай у ранньому дитячому (іноді у грудному) віці. Напади виникають у період відпочинку після важкого м'язового навантаження. Швидко розвивається слабкість, яка може тривати протягом кількох годин. Вона особливо відчувається у ногах; функцію дихання зазвичай не порушено. Часто адинамії супроводжує міотонія, що зберігається між нападами, що найбільш яскраво проявляється у вигляді запізнювання руху повік при погляді вниз (див. рис. 21-8, б).
Під час нападу рівень калію у сироватці часто підвищений, проте для того, щоб визначити це з достовірністю, можуть знадобитися багаторазові дослідження під час кількох нападів. Штучно спровокувати напад можна за допомогою калієвого навантаження (2-3 г всередину), проте його слід проводити лише під контролем ЕКГ. Повторні напади усувають діакарбом. Тяжкі форми захворювання характеризуються розвитком хронічної, слабо вираженої слабкості та дистрофічними змінами у м'язах.
Гіпокаліємічний пароксизмальний параліч. Сімейний пароксизмальний параліч, який також успадковується за домінантним типом, особливо важко протікає у хлопчиків. На відміну від гіперкаліємічної форми перший напад з'являється у пізньому дитячому чи ранньому підлітковому віці. Причиною є вживання багатої їжі, багатої на вуглеводи, або відпочинок після фізичного навантаження. Зазвичай напад починається наступного ранку після важкого фізичного навантаження і рясної вечері. Він характеризується м'язовою слабкістю та арефлексією. Можливо порушена функція дихання. Може приєднатися аритмія, включаючи шлуночкову екстрасистолію та тахікардію. Приступи можуть тривати більше 24 год. У паралітичній фазі рівень калію в сироватці зазвичай знижується (2-3 ммоль/л). Основний дефект невідомий. У хворих з повторними тяжкими нападами розвиваються хронічна м'язова слабкість та патологічні зміни у м'язах. Лікування під час нападів полягає у прийомі калію хлориду; його початкова доза становить 2-3 г. Діакарб сприяє зменшенню частоти нападів.
Пароксизмальна міоглобінурія (ідіопатична міоглобінурія). Ідіопатична міоглобінурія є різнорідною групою захворювань, при яких напади паралічу з міоглобінурією виникають спонтанно або після інтенсивного фізичного навантаження. Захворювання успадковується за домінантним типом, зчеплене з Х-хромосомою. М'язи, найчастіше литкові та стегнові, під час нападу стають болючими та припухлими. Сеча набуває темно-червоного або коричневого кольору. Міоглобінурія може зумовити некроз ниркових канальців, що призводить до летального результату в результаті ниркової недостатності.
Діагноз підтверджується виявленням у сечі міоглобуліну. Позитивна бензидинова проба за відсутності в сечі еритроцитів підтверджує присутність у ній міоглобіну, особливо якщо сироватці не визначається гемоглобін. Гемоглобін визначають за допомогою спектрофотометрії. Пароксизмальну міоглобінурію слід відрізняти від хвороби Мак-Ардла, недостатності карнітинпальмітілтрансферази та міоглобінурії після незвичного інтенсивного фізичного навантаження або травми м'язів у здорової людини. Міоглобінурія після тяжкого м'язового навантаження зустрічається при псевдогіпертрофічній м'язовій дистрофії (хвороба Дюшенна).
Лікування полягає у дотриманні постільного режиму; за необхідності проводять штучну вентиляціюлегенів. Для запобігання нирковій недостатності необхідно призначати хворому на рясне пиття.
Недостатність карнітинпальмітілтрансферази.При дефіциті цього ферменту порушується перенесення довголанцюгових жирних кислот у мітохондріальні сегменти, в яких здійснюються окислення та продукція кетонів. Недостатність ізоферменту II типу успадковується за рецесивним типом. Внаслідок його дефіциту порушується кетогенез у тканинах, у тому числі у м'язовій та печінковій. Перші ознаки хвороби з'являються частіше у дітей шкільного та підліткового віку. Вони полягають у повторних епізодах болів у м'язах, слабкості та підвищенні температури тіла після фізичного навантаження чи голодування. Міоглобінурія, що супроводжує напади, може призвести до ниркової недостатності. Голодування призводить до гіпоглікемії. Між нападами діти виглядають здоровими. Захворювання необхідно диференціювати від інших станів, що супроводжуються періодичними слабкістю та міоглобінурією. Диференціально-діагностичну цінність має метод визначення активності карнітинпальмітілтрансферази. Вона знижується в м'язовій та печінковій тканинах, лейкоцитах та культурі фібробластів. Дотримання дієти, що складається з продуктів, збагачених вуглеводами та знежирених, сприяє зменшенню кількості нападів.
М'язові дистрофії. Ці аномалії належать до групи сімейних захворювань, що супроводжуються дегенерацією м'язових волокон. Класифікація м'язових дистрофій ґрунтується на таких ознаках, як час початку, швидкість прогресування, розподіл уражень по групах м'язів та тип спадкування.
Псевдогіпертрофічна м'язова дистрофія. Дитяча, або форма Дюшенна, є найбільш поширеною формою м'язової дистрофії; її частота становить 0,14 на 1000 дітей. У класичній формі вона протікає тільки у хлопчиків, причому успадкування, зчеплене з Х-хромосомою, відбувається приблизно у 50% пробандів. В інших випадках захворювання обумовлено новими мутаціями. Повідомляється про рідкісну форму м'язової дистрофії, по клініці ідентичної формі Дюшенна, але успадкованої за рецесивним типом з однаковою частотою захворювання хлопчиків та дівчаток. Достовірно діагностувати захворювання рідко можливо у дитини до 3 років. В анамнезі зазвичай є вказівки на те, що у дитини був уповільнений розвиток рухових функцій, він пізно почав сидіти, ходити та бігати, що, природно, свідчить про більш ранньому початкузахворювання. Перевалюється (качина) хода, труднощі при підйомі сходами, гіпертрофія литкових м'язів відносяться до звичайних клінічних проявів. У деяких випадках до процесу залучаються також інші м'язи, зокрема дельтоподібні, плечопроменеві, м'язи язика.
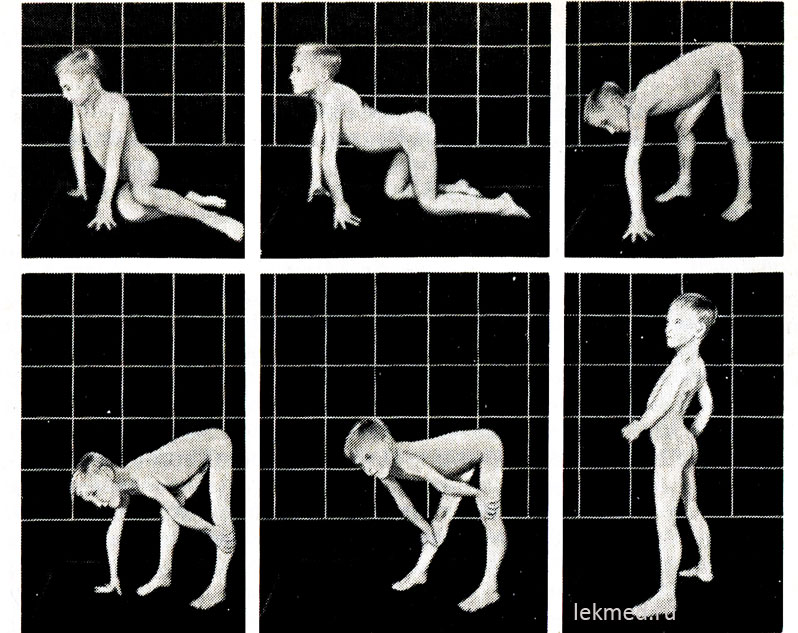
Рис. 21-9. Типові пози, що приймаються під час вставання з статі (симптом Говерса) дитиною віком 7 років із псевдогіпертрофічною міопатією.
У положенні стоячи (остання фотографія) відзначається значно виражений лордоз.
На початку захворювання гіпертрофовані м'язи мають значну силу, але пізніше вона зменшується (псевдогіпертрофія), так як збільшення маси м'язів відбувається за рахунок їх жирової інфільтрації. Сила гіпертрофованого литкового м'яза значно перевищує силу м'язів передньої поверхні ноги, що пояснює часті контрактури сухожилля п'яти і ходіння дитини на пальцях. Слабкість м'язів тазового пояса виражається в характерній качині (лордична) ході та утрудненнях, які дитина відчуває, коли встає зі становища сидячи на підлозі. При досить тяжких формах м'язової дистрофії у дитини відзначається симптом Говерса: встаючи з підлоги, він насамперед стає на коліна, спираючись на руки, а потім піднімається, послідовно відштовхуючись руками від гомілок, колінних суглобів та стегон (рис. 21-9). Визначити слабкість м'язів плечового пояса можна, утримуючи дитину в піднесеному положенні за пахви. У нормі він намагається утриматися, притискаючи руки до тулуба; при м'язовій дистрофії він ніби прослизає через руки обстежуючого. Хвора дитина часто не може підняти руки над головою. У пізні стадії захворювання розвивається значна м'язова атрофія. Зазвичай до віку 12 років дитина вже не може ходити. Хворі у 75% випадків помирають у віці до 20 років. У більшості з них відзначається кардіоміопатія, яка в деяких випадках є причиною раптової смерті. Якщо спадкування зчеплене з Х-хромосомою, а захворювання почалося у старшому дитячому віці, тривалість життя залишається великою (м'язова дистрофія Беккера). Середній коефіцієнт розумового розвитку в дітей із формою Дюшенна становить 80; у 25% дітей відзначається відставання розумового розвитку.
При диференціальної діагностиким'язової дистрофії Дюшенна слід мати на увазі хворобу Вердніга-Гоффмана у дітей старшого грудного віку і такі м'язові захворювання, як ендокринні міопатії, недостатність карнітину, хвороби накопичення глікогену та поліміозиту. Іноді при контрактурах сухожилля п'яти і ходженні дитини на пальцях ніг можна припустити церебральний параліч, проте при м'язовій дистрофії відсутні характерні для церебрального паралічу спастичність і гіперрефлексія.
Діагноз заснований на визначенні активності сироваткових ферментів, даних електроміографії та біопсії м'язової тканини. Активність ферментів, особливо креатинфосфокінази, ще до розвитку клінічних симптомівчасто перевищує норми у 10 разів навіть у дітей грудного віку. На електроміограмі виявляють насамперед зменшення тривалості та зниження амплітуди рухових потенціалів. Гістологічні зміни полягають у дегенерації м'язових волокон. Вони часто розрізняються за розміром і частково заміщені жиром та сполучною тканиною. Розмір їх ядер також змінюється. Діагноз можна встановити при народженні шляхом визначення активності креатинфосфокінази. Методи виявлення жінок-носіїв ще не розроблені, незважаючи на те, що у 60-80% з них виявляють незначне або помірне підвищення її рівня. Ці ознаки типові більше дитячого віку, ніж наступних періодів життя.
Ефективних методівлікування не існує. Слід якнайбільше сприяти підтримці активного стану хворого та його здатності ходити. Необхідно стежити за тим, щоб дитина уникала інтенсивного фізичного навантаження, оскільки вона може бути причиною розриву м'язових волокон. У деяких випадках хірургічне подовження сухожилля п'яти сприяє можливості ходити, проте тривалий постільний режим після ортопедичної корекції може посилити м'язову атрофію. Важливу роль грає генетичне консультування.
Уроджена м'язова дистрофія. Захворювання успадковується за аутосомно-рецесивним типом і характеризується м'язовими гіпотонією та слабкістю у дитини грудного віку. Воно входить до групи станів, визначених як «млява дитина» (див. табл. 21-1). Початок захворювання відноситься до внутрішньоутробного періоду. Іноді у новонародженого відзначають виражену атрофію м'язів, їхню контрактуру, обмежену рухливість суглобів. Диференціювання від хвороби Вердніга-Гофманна утруднене. Фасцикуляції мови, характерні останньої, відсутні при м'язової дистрофії. Сухожильні рефлекси пригнічені, але не втрачені. У процес залучаються м'язи, що у диханні, включаючи діафрагму. При тяжкому перебігу смерть настає у віці до 1 року внаслідок дихальної недостатності; при легших формах нормальна життєздатність зберігається протягом багато часу. Підвищення активності сироваткових ферментів не відзначають, хоча у м'язах відбуваються дистрофічні зміни.
Плечолопаточно-лицьова форма м'язової дистрофії. Ця досить легка формам'язової дистрофії успадковується за аутосомно-домінантним типом. Починається вона зазвичай у віці 10-20 років і характеризується слабкістю та атрофією м'язів обличчя та плечового поясу. Особа повністю амімічна, хворий не може заплющити очі і зробити свист. Захворювання прогресує повільно та сумісне з нормальною тривалістю життя. Діагноз заснований на клінічних даних та типі успадкування. Результати біопсії м'язової тканини свідчать про дистрофічні зміни в ній. Рівень креатинфосфокінази у сироватці може залишатися в межах норми або трохи підвищується.
Тазова форма м'язової дистрофії. Ця група неоднорідних порушень характеризується повільним прогресуванням м'язової дистрофії та успадковується за аутосомно-рецесивним типом. Початок захворювання відноситься до старшого дитячого, підліткового або дорослого віку. Зазвичай уражаються м'язи тазового пояса.
Очна форма міопатії. Дистрофічні зміни відбуваються переважно у зовнішніх очних м'язах. Починається захворювання у дитячому чи підлітковому віці. При ньому прогресують птоз та обмеження рухів очних яблук. Іноді слабкість поширюється на м'язи обличчя та шиї. Захворювання слід диференціювати від міастенії та паралічів черепних нервівпри пухлинах стовбура мозку.
Прогресуюча офтальмоплегія, що починається у дитячому чи підлітковому віці, пов'язана з атиповою пігментною дегенерацією сітківки та блокадою серця (синдром Кернса – Сейєрса). Зазвичай з нею пов'язані і прогресуюча атаксія, відставання росту та статевого дозрівання. Під сарколемою м'язів визначають великі скупчення атипових мітохондрій. Генетична природа цього процесу встановлено. Можна контролювати можливість раптової смерті від порушення серцевої провідності за допомогою кардіостимулятора.
Міотонічна дистрофія. Незважаючи на те, що міотонічна дистрофія починається ніби у дорослої людини, її початок все частіше реєструють у дітей грудного та пізнішого дитячого віку. Вона успадковується за аутосомно-домінантним типом. Її початок у дитячому віці свідчить про те, що на міотонію страждає мати. Відповідно до цього внутрішньоутробні фактори можуть впливати на вираженість захворювання у дитини. Вже на момент народження він може визначатися м'язова гіпотонія, в нього відсутня здатність смоктати. Відставання фізичного та розумового розвитку зазвичай виявляється пізніше. У ранньому дитячому віці м'язові слабкість та атрофія поширюються в основному на лицьові, щелепні та скроневі м'язи. Зазвичай відзначається двосторонній птоз. До діагностично значимих методів належать перкусія м'язів, електроміографія; типова для цих хворих нездатність розтиснути стиснуту в кулак кисть (див. Вроджена міотонія). Слабкість та атрофію м'язів кінцівок та тазового поясу (зазвичай дистальні групи) виявляють у старшому дитячому чи підлітковому віці. У дорослих цього захворювання супроводжують катаракта, облисіння, атрофія яєчок.
Діагноз заснований на виявленні ознак міотонії, характерному розподілі м'язової слабкості, успадкування за домінантним типом, дистрофічних змін м'язів. У дитячому віці перебіг захворювання може бути несприятливим, часто супроводжує розумова відсталість. До юнацького віку м'язова слабкість виступає передній план. При функціональних порушеннях показано лікування новокаїном та хінідином.
Крім хворобливих порушень, отриманих при травмах (наприклад, розривів та розтягувань), порушення у м'язах можуть бути і за відсутності зовнішніх факторів впливу. До хвороб м'язів можна віднести:
М'язова судома;
Ревматичні захворювання;
Запалення;
Генетичні захворювання;
Порушення обміну речовин;
Зміна м'язових клітин.
Розглянемо все захворювання докладніше.
М'язова судома
Судома може виникнути внаслідок зневоднення організму (ексикозу). У цей момент м'язи скоротилися та стали твердими, потім вони повільно розслаблюються. Судома може виникнути вночі чи вранці. Людина різко починає відчувати сильний більу м'язі. Судоми найчастіше зустрічаються у людей похилого віку. Коли на м'язи йде занадто велике навантаження або їх порушується, то з'являються затвердіння. М'язові волокна перетворюються на м'язову тканину, у якій промацуються тверді ділянки як вузлів. У таких випадках необхідно пити багато рідини, щоб відновити водно-сольовий баланс в організмі. Також на допомогу приходить масаж. Якщо болі в м'язах не припиняються, необхідно видатися лікарю. Лікують затвердіння за допомогою масажу, вітаміну Е та теплих ванн.
Ревматичні захворювання
Є дуже велика кількість захворювань, які можна віднести до ревматичних. При цих захворюваннях джерело ураження - сам м'яз, або кровоносні судини, які живлять м'яз. З'являються болі в стегнах та плечах. Деякі захворювання ревматичного характеру (наприклад, дерматоміозити) вражають м'язи. В даному випадку необхідне лікування гормонами – глюкокортикоїдами. Вони пригнічують запалення, але викликають побічні ефекти. Тому ревматичні захворювання намагаються придушити за допомогою протизапальних ліків або фізіотерапії.
Порушення гормонального характеру
Болюча слабкість м'язів у медицині називається як ендокринна міопатія, яка виникає внаслідок посилення функції щитовидної залози або надниркових залоз. Після лікування біль зникає.
Запалення м'язів
Запалення м'язів називається міозит. Симптоми цього захворювання такі ж, як і при ревматизмі, але відмінною рисоює запалення самих м'язів. Для міозитів властиві болі та яскрава м'язова слабкість. Лікують запалення м'язів так само, як і ревматичні захворювання.
Нестача мінеральних речовин
Для нормального функціонування м'язів потрібні певні речовини. При дефіцит калію виникає параліч. Особливо це відчувають молоді люди та дітки вранці після тяжкого минулого дня. Лікують за допомогою препаратів, які містять калій. Крім того, перед сном не варто багато їсти та активно займатися спортом.
Нестача ферментів
Діти можуть рідко спостерігатися дефіцит ферменту. Часто зустрічаються порушення функцій ферментів, що беруть участь у розщепленні глюкози та глікогену, які є джерелом енергії для м'язів. В результаті вродженого дефіциту ферменту м'язи одержують мало енергії внаслідок ослаблення їхньої роботи. Людина з таким діагнозом має уникати фізичного навантаження.
Болюча втома м'язів
Втома м'язів, що супроводжується болем, з'являється внаслідок ацидозу. Для отримання енергії при високих навантаженнях відбувається розщеплення глюкози до молочної кислоти, яка важко виводиться з організму. Нагромаджуючись у м'язах, молочна кислота викликає хворобливі відчуття.
У всьому світі спортсмени для запобігання болю в м'язах, поліпшення харчування, відновлення та лікування вживають сік із мангустину.
Необхідно вживати чисту воду.
Прогресують дегенеративні захворювання нервової системивиникають у результаті генетично детермінованої патології чи дефекту ембріонального розвитку. Загальними проявами цих захворювань є: дегенеративний характер і системність ураження нервової тканини, прогресуючий перебіг. До них відносяться, зокрема, сирингомієлія, при якій у спинному мозку формуються довгі порожнини, що руйнують задні роги. Це призводить до дефекту больової та температурної чутливості, атропатій.
Група спадкових атаксій досить численна, їх основним проявом є атаксія, пов'язана з патологією мозочкових шляхів або глибокої чутливості.
Бічний аміотрофічний склероз (БАС)- Найважче і швидко прогресуюче захворювання, сопронних шляхів у спинному мозку. При цьому виникає комбінація з атрофічного парезу і пірамідної симптоматики, тобто рухові розлади, що розвиваються, носять характер одночасно як периферичного, так і центрального паралічу.
Хвороба Паркінсона є прогресуючим захворюванням, в основі якого лежить первинне ураження дофамінергічних нейронів, що містять пігменти, щільної частини чорної субстанції та інших пігментовмісних ядер стовбура. Ризик для найближчих родичів хворого приблизно 10 разів вище, ніж у популяції. Для хвороби Паркінсона характерна тріада симптомів: тремор, підвищення м'язового тонусу та гіпокінезія; Критерії діагнозу дуже складні. Клініка хвороби виникає лише тоді, коли гине понад 80% нейронів. У ряді випадків хвороба дебютує у віці до 18 років (так званий юнацький паркінсонізм), здебільшого – у старшому віці. Це висуває підвищені вимоги до своєчасності та адекватності лікування.
Сучасна адекватна терапія надає багато можливостей для хорошої компенсації порушених функцій, і навіть збереження чи відновлення соціальної адаптації.
Спадкові нервово-м'язові захворювання
Дегенеративні нервово-м'язові захворювання- Захворювання з переважним ураженням нервово-м'язового апарату спадкового характеру. Вони позначаються також як прогресуючі нервово-м'язові дистрофії (ПМД) та становлять найзначнішу групу серед усіх спадкових захворювань.
Класифікація прогресуючих м'язових дистрофій
- Прогресують м'язові дистрофії - генетично детерміновані розлади з первинною прогресуючою дегенеративною зміною м'язів (без первинної патології периферичного мотонейрону). За них саме м'язова тканинає мішенню первинного генного дефекту, через який відбувається аномальний синтез м'язового білка мйодистрофіну та прискорюється його розпад. Поразка нервової системи при міопатії має вторинний характер.
- Спінальні аміотрофії - первинне генетично детерміноване ураження передніх рогів спинного мозку з вторинним прогресуючим периферичним паралічем та атрофією м'язів.
- Невральні аміотрофії – первинні генетично детермінований синдром поліневропатії (внаслідок мієлінопатії) з розвитком вторинної аміотрофії та вегетативно-сенсорних порушень.
Первинні прогресуючі м'язові дистрофії
Різні форми захворювань проявляються у різному віці – від 1-2 років до 40-50 і старше. Вони характеризуються руховою незручністю, нестійкістю, падіннями під час ходьби, стомлюваністю. У дитини виникає страх та небажання ходити. У хворих із сформованою ходою виникає « качина хода»- перевалку.
Для деяких форм характерна псевдогіпертрофія м'язів, що частіше уражаються литкові м'язи: їх атрофія з маскуванням атрофії і навіть збільшенням розміру через розростання сполучної жирової тканини. Слабкість та атрофії м'язів спочатку локалізовані у м'язах тазового поясу, з максимальною вираженістю у проксимальних відділах ніг.
Є виражений поперековий лордоз, сколіоз, «крилоподібні» лопатки, вузька «осина» талія. Підйом із становища сидячи утруднений і діти вдаються до допоміжних прийомів (прийоми Говерса) - «підбір драбинки», «підбір по собі». Відомі випадки з недостатнім розвитком деменції. Страждає серцевий м'яз. Далі хворі втрачають здатність самостійно ходити. У процес залучається серцево-судинна система(Розвивається дилятаційна або гіпертрофічна кардіоміопатія).
Вторинні - спинальні та невральні м'язові дистрофії.
Спинальні м'язові дистрофії (аміотрофії) успадковуються за аутосомно-рецесивним типом. Ген спинальної м'язової атрофії картирован на хромосомі 5gl1.2-13.3.
Можуть виявлятись ранні ознаки бульварних розладів. Затримка моторного розвитку. Під час проведення електроміографії виявляється ураження передніх рогів спинного мозку. Перебіг захворювання прогресує.
При спінальних формах аміотрофії на електромі-ограмі у спокої реєструються потенціали фібриляцій; швидкість поширення імпульсу нервами кінцівок щодо збережена, але може і знижуватися в результаті загибелі спинальних мотонейронів.
Найбільш частим варіантом невральної аміотрофії є невральна аміотрофія Шарко-Марі-Тута. Вона за своїми клінічними проявами нагадує сенсомоторну поліневропатію, що дистально акцентована і починається зі стоп і гомілок. Тече доброякісно, повільно. Згодом формується характерна деформація ніг - на кшталт «ніг лелеки» або «галіфе»: тонкі внаслідок атрофії гомілки при збережених м'язах стегон. Спочатку «випадають» ахіллові рефлекси, потім знижуються колінні.
При електронейроміографії реєструється грубе зниження швидкості поширення імпульсу нервами кінцівок.
Міастенія. Міастенічний та холінергічний кризи
Міастенія(miastenia gravis pseudoparalitica) - важке нервово-м'язове захворювання аутоімунної природи, що характеризується патологічною стомлюваністю та слабкістю поперечно-смугастої мускулатури (Акімов Г. А., Одинак М. М., 2000).
Етіопатогенез. Основна ланка - виникнення аутоантитіл до нікотинових холінорецепторів кінцевої пластинки м'язового волокна та блок нервово-м'язової передачі. Є зв'язок патогенезу міастенії з ураженням вил очкової залози. Часто виявляється тимома (до 40% випадків), рідше – атрофія тимусу.
Клініка. Міастенія може виникнути у будь-якому віці, але частіше – між 16 та 40 роками, проте бувають і більш ранні, і пізніші форми (відзначені піки захворюваності у 30 та 70 років). Жінки хворіють частіше за чоловіків. Основний симптом - патологічна стомлюваність м'язів з розвитком їх наростаючої слабкості при повторних рухах, наприклад, виникнення двоїння або птозу під час читання.
М'язова слабкість, що розвивається при міастенії, відрізняється від периферичних або центральних парезів тим, що при повторенні рухів, особливо в швидкому темпі, вона різко зростає і може досягати ступеня повного паралічу. Після відпочинку, сну, перші рухи можуть бути нормальними, проте за наступних з'являється втома, ступінь якого прогресує при продовженні навантаження.
Міастенічний епізод може розвинутися у новонароджених дітей, які з'явилися на світ від матерів, які страждають на міастенію (так звана міастенія новонароджених). Ступінь компенсації рухових розладів може бути повним, достатнім (для самообслуговування в побуті), поганим (необхідний сторонній догляд). Найбільш грізне ускладнення міастенії – міастенічний криз.
Міастенічний криз- невідкладний критичний стан, що раптово розвивається, в результаті блоку нервово-м'язової провідності. Основні симптоми - генералізована м'язова слабкість, що швидко розвивається, доходить до ступеня тетраплегії.
Ускладнення:
- порушення дихання при бульбарній формі,
- ризик обтурації дихальних шляхівгустою мокротою, що накопичується,
- можливість аспірації їжі або «клапанної асфіксії» через заходження мови та слабкість надгортанника,
- виключення діафрагми та слабкість міжреберних дихальних м'язів.
Передозування антихолінестеразних препаратів може призвести до розвитку холінергічного кризу з різким погіршенням самопочуття. Невідкладна допомога» виявляється медичними працівникамиу реанімаційному відділенні чи блоці (палаті) інтенсивної терапії.
Запалення м'язів – захворювання, основними симптомами яких виступає м'язова слабкість, пов'язана із запаленням поперечнополосатої мускулатури. До запалень м'язів відносять ідіопатичні запальні міопатії, міопатії, пов'язані з інфекцією, та міопатії, пов'язані з впливом ЛЗ та токсинів. Серед них найбільш важливими є ознаки поліміозиту та дерматоміозиту. У цій статті ми розглянемо симптоми запалення м'язів та основні ознаки запалення м'язів у людини. Крім того, ми розповімо про діагностику запалених м'язів.
Симптоми запалення м'язів
У дебюті симптомів запалення м'язів більшість хворих відзначають ознаки нездужання, загальної слабкості, ураження шкіри (при дерматоміозіті). Надалі поступово (протягом кількох тижнів) до запалення м'язів приєднується симптоми прогресуючого наростання слабкості у проксимальних групах м'язів. У деяких пацієнтів з ознаками запалення м'язів (дітей та осіб молодого віку) спостерігають гострий початок, що часто поєднується з вираженими конституційними ознаками (лихоманка, схуднення та ін) і міалгіями.
Дуже повільне (протягом кількох років) наростання м'язової слабкості при симптомах запалення м'язів спостерігають частіше у літніх хворих, які страждають на міозит з "включеннями". Вкрай рідко розвивається так званий аміотрофічний дерматоміозит при запалених м'язах, при якому основною ознакою дуже довго виступає типове ураження шкіри. У хворих з антисинтетазним синдромом ранніми ознакамизапалення м'язів можуть бути феномен Рейно, поліартралгії або поліартрит та задишка, обумовлена інтерстиціальним легеневим фіброзом.
Симптоми ураження м'язів при запаленні
Ведучий клінічна ознаказапалення м'язів - симетрична слабкість проксимальних груп м'язів верхніх та нижніх кінцівок, а також м'язів, що беруть участь у згинанні шиї. Це призводить до утруднення при підйомі з низького випорожнення, посадці в транспорт, при вмиванні та зачісуванні. Хода при симптомах запалення м'язів стає незграбною, шкутильгаючою, хворі не можуть піднятися без сторонньої допомоги і відірвати голову від подушки. Запалення м'язів глотки, гортані та стравоходу призводить до дисфонії, утруднення ковтання, нападів кашлю. Ознаки ураження дистальної мускулатури виникають рідко (10%), виражене меншою мірою, ніж ураження проксимальної мускулатури, і виявляється головним чином при міозиті з "включеннями". У половини хворих з симптомами запалення м'язів можливі міалгії або болючість м'язів при пальпації, набряк м'язів, але м'язові атрофії розвиваються тільки у хворих, які тривалий час страждають на поліміозит/дерматоміозит, особливо без адекватної терапії. М'язова гіпертрофія характерна ознакадля м'язових дистрофій і не спостерігається при поліміозит/дерматоміозит.
Симптоми ураження шкіри при запаленні м'язів
Патогномонічна ознака дерматоміозиту при запаленні м'язів. Шкірні ознаки включають еритематозний (геліотропний) висип, що локалізується на верхніх віках, вилицях, крилах носа, в області носогубної складки, в зоні "декольте" і на верхній частині спини, над ліктьовими та колінними, п'ястно-фаланговими та проксимальними міжфаланговими суглобами, на волосистій частині голови. Злегка піднімаються або плоскі еритематозні висипання, що лущиться, локалізуються над суглобами пальців кистей, отримали назву "ознака Готтрона" при запаленні м'язів. Характерні шкірні ознаки, що спостерігаються не тільки при дерматоміозиті, але і при поліміозиті: почервоніння, лущення та розтріскування шкіри долонь ("рука механіка або ремісника"), гіпертрофія кутикули, навколонігтьова еритема, телеангіектазії. При капіляроскопії судин при запаленні м'язів навколонігтьового ложа відзначають розширення та дилатацію капілярних петель, частіше при перехресному синдромі, рідше при дерматоміозіті. Фотодерматит та шкірний свербіжвідзначають рідше.
Симптоми ураження суглобів при запаленні м'язів
Симптоми ураження суглобів часто передує розвитку м'язової патології при запаленні м'язів. Найчастіше залучаються дрібні суглоби кистей, променево-зап'ясткові суглоби, рідше - ліктьові та колінні суглоби. Поразка двостороння симетрична, нагадує таку при ревматоїдний артрит, як правило, має тимчасовий характер, симптоми запалення м'язів швидко купіруються при призначенні глюкокортикоїдів. Однак описано розвиток хронічного артриту, що деформує, з підвивихами суглобів кистей, але без ерозивних змін за даними рентгенологічного дослідження.
Симптоми кальцинозу при запаленні м'язів
Ознаки кальцинозу виникають на пізніх стадіях, частіше при ювенільному дерматоміозіті Кальцифікати локалізуються підшкірно або в сполучній тканині навколо м'язових волокон, часто в зонах мікротравматизації над ліктьовими та колінними суглобами, на згинальних поверхнях пальців та сідницях.
Симптоми ураження легень при запаленні м'язів
Провідною клінічною ознакою запалення м'язів виступає експіраторна задишка, яка може бути пов'язана з ураженням діафрагмальних м'язів, розвитком серцевої недостатності, інтеркурентною легеневою інфекцією, токсичною поразкоюлегень, пов'язаних із прийомом деяких препаратів, наприклад метотрексату. Описано розвиток симптомів гострого дифузного альвеоліту, що виходить на перший план клінічній картинізапалення м'язів і проявляється непродуктивним кашлем та швидкопрогресуючою дихальною недостатністю. Найчастіше спостерігають повільний прогрес інтерстиціального легеневого фіброзу, у деяких хворих виявляється тільки при спеціальному обстеженні. У найважчих випадках розвивається аспіраційна пневмонія.
Симптоми ураження серця при запаленні м'язів
Ознаки ураження серця при поліміозит/дерматоміозит у більшості випадків протікає безсимптомно. Іноді при спеціальному обстеженні виявляють симптоми порушення ритму та провідності (тахікардію, аритмію). Застійна серцева недостатність, пов'язана з дилатаційною кардіоміопатією, розвивається рідко. Феномен Рейно частіше спостерігають при дерматоміозиті, антисинтетазному синдромі та у хворих з перехресним синдромом поліміозиту/дерматоміозиту з системними захворюваннямисполучної тканини.
Ознаки інших судинних порушень при запаленні м'язів
Описані інфаркти навколонігтьового ложа, петехії, livedo reticularis (гіллястий малюнок на шкірі кінцівок та тулуба). Ураження нирок спостерігають рідко, хоча можливий розвиток протеїнурії та навіть нефротичного синдрому. Виражена міоглобінурія може наводити кОПН.
Ознаки запалення м'язів
Основне значення у патогенезі поліміозиту/дерматоміозиту мають клітинні імунні реакції. При імунопатологічному дослідженні ураженого м'яза виявляють інфільтрацію Т- та В-лімфоцитами та макрофагами, що знаходяться в активованому стані. При цьому Т-клітини мають цитотоксичну активність щодо міофібрил. Між поліміозитом та дерматоміозитом виявлено ознаки певних імунопатологічних відмінностей. При дерматоміозиті у складі м'язового інфільтрату переважають СЕ4+-Т-лімфоцити, макрофаги та В-лімфоцити, а при поліміозиті – цитотоксичні СЕ8+-Т-лімфоцити. Припускають, що при ознаках дерматоміозиту розвивається гуморальна імунна відповідь, що призводить до активації комплементу, що вражає внутрішньом'язові мікросудини, а при поліміозіті переважають клітинні цитотоксичні реакції, опосередковані СЕ8+-Т-лімфоцитами, що синтезують цитотоксин. Патогенетичне значення міозитспецифічних аутоантитіл при запаленні м'язів не доведено.
Причини появи симптомів запалення м'язів
Причини виникнення запалення м'язів точно не з'ясовані. На роль інфекційних факторів побічно вказує частіший початок захворювання взимку і ранньою весною (особливо у хворих на ювенільний дерматоміозит), що за часом збігається з епідеміями інфекцій. Про участь генетичної схильності свідчить можливість розвитку поліміозиту/дерматоміозиту у монозиготних близнюків та кровних родичів хворих. Носіння деяких Аг головного комплексу гістосумісності (HLA) більш тісно пов'язане не із самим запаленням м'язів, а з певними імунними порушеннями, в першу чергу з гіперпродукцією міозитспецифічних аутоантитіл.
Поширеність ознак запалення м'язів
Захворюваність на запалення м'язів у популяції коливається від 2 до 10 випадків на 1 млн населення на рік. Залежно від віку спостерігають два піки захворюваності: у 5-15 років (ювенільний дерматоміозит) та 40-60 років. Переважна стать жіноча (співвідношення кількості хворих жінок і чоловіків становить 2-3:1)
Діагностика запалення м'язів
Загальний аналізкрові при запаленні м'язів: характерних ознак немає, збільшення ШОЕ спостерігають рідко, головним чином розвитку системних проявів.
Біохімічні аналізи крові при діагностиці запалених м'язів
Загальноприйнятий показник пошкодження скелетної мускулатури - КФК, збільшення якої при поліміозит/дерматоміозит відрізняється більш високою чутливістюта специфічністю порівняно з іншими лабораторними тестами. Збільшення КФК при запаленні м'язів у різні періоди хвороби буває у 95% хворих на поліміозит/дерматоміозит. Концентрація КФК може зростати до появи клінічних ознак запалення м'язів загострення поліміозиту/дерматоміозиту, а рівень може знижуватися до розвитку клінічного поліпшення. Іноді у хворих рівень КФК може бути в межах норми, незважаючи на тяжке ушкодження м'язів за даними морфологічного дослідження, у цьому випадку показник не корелює з динамікою клінічних та морфологічних ознак активності. Необхідно мати на увазі, що нормальний рівень КФК може спостерігатися у хворих з тяжкою атрофією м'язів на пізніх стадіях хвороби, в дебюті дерматоміозиту і при симптомах пухлинного міозиту.
Збільшення МВ-фракції КФК спостерігають при ознаках поліміозиту/дерматоміозиту без некрозу міокарда. Збільшення активності трансаміназ не специфічне ураження скелетної мускулатури. У деяких хворих із генералізованою слабкістю ізольоване збільшення трансаміназ змушує запідозрити гепатит.
Імунологічна діагностика запалених м'язів
До міозитспецифічних AT відносять AT до аміноацилсинтетазів транспортної РНК (антисинтетазні AT), в першу чергу AT до гістиділу тРНК синтетази (Jo-1). AT Jo-1 виявляють у половини хворих на поліміозит/дерматоміозит, тоді як інші антисинтетазні AT вкрай рідко (5%). Продукція антисинтетазних AT асоціюється з розвитком так званого антисинтетазного синдрому, що характеризується гострим початком, інтерстиціальним ураженням легень, лихоманкою, симетричним артритом, феноменом Рейно, ураженням шкіри кистей типу "руки механіка" при запаленні м'язів.
Інструментальні методивизначення запалення м'язів
Електроміографія для діагностики запалення м'язів – чутливий, але неспецифічний метод діагностики запальних міопатій. До типових симптомів, що спостерігаються більш ніж у 90% хворих при дослідженні проксимальних і параспінальних м'язів, відносять ознаки патологічної спонтанної активності міофібрил (потенціали фібриляції, складні повторювані розряди та ін) при роздратуванні і в спокої, короткі низькоамплітуди. Нормальна електрична активністьпри електроміографії у більшості випадків дозволяє виключити діагноз поліміозиту/дерматоміозиту. Електроміографія - корисний методконтролю за ефективністю лікування запалення м'язів, особливо при сумнівних результатівлабораторних та клінічних досліджень. Однак дані електроміографії погано корелюють з клінічними проявамим'язової слабкості. Важливо, що з стероїдної міопатії спостерігають такі самі (хоча й менш виражені) зміни, як і за активному міозиті.
Біопсія м'язів при симптомах запалення використовується для підтвердження діагнозу, навіть за наявності характерних клінічних, лабораторних та інструментальних ознак запалення м'язів. Найбільш інформативна біопсія м'яза, залученого до патологічного процесу, але без вираженої атрофії.
Рентгенологічні дослідження для діагностики запалення м'язів. Рентгенологічні симптоми запалення суглобів не характерні. При рентгенологічному дослідженні легень часто виявляють ознаки базального пневмосклерозу та інтерстиціального легеневого фіброзу. Більш чутливим методом вважають рентгенівську КТ з високою роздільною здатністю (РКТ).
ЕКГ під час діагностики ознак запалення м'язів. Для раннього виявлення прогностично несприятливих порушень ритму та провідності доцільно проведення добового моніторування ЕКГ (за Холтером).
Симптоми міозиту м'язів
Міозит із "включеннями" - ознака запалення м'язів
Серед хворих із симптомами запалення м'язів частота міозиту із "включеннями" коливається від 15 до 28%. До його ознак відносять такі:
- літній вікхворих (середній вік близько 60 років);
- частіша поразка чоловіків (співвідношення чоловіків і жінок 2:1);
- дуже повільний розвиток слабкості та атрофії у проксимальних, а й у дистальних групах м'язів - характерний ознака при запаленні м'язів;
- асиметричність ураження;
- нормальне чи помірне підвищення активності КФК;
- рідкісна асоціація із системними захворюваннями сполучної тканини та злоякісними новоутвореннями;
- відсутність міозитспецифічних та інших аутоантитіл - характерна ознака при запаленні м'язів;
- резистентність до глюкокортикоїдів та інших методів фармакотерапії (особливо характерна ознака).
Характерна морфологічна особливість - амілоїдогенні "окреслені" васкуолі, великі внутрішньоядерні та внутрішньоцитоплазматичні "включення" (при світловій мікроскопії) та мікротубулярні елементи, що виявляються при електронної мікроскопії. Певне діагностичне значеннямає електроміографія, при якій виявляють змішані міопатичні та невропатичні ознаки.
Симптоми міозиту, асоційованого із системними захворюваннями сполучної тканини.
Проксимальна м'язова слабкість – частий клінічний ознака системних запалень м'язів, а ознаки інших хвороб цієї групи виявляють приблизно у 20% хворих на поліміозит/дерматоміозит. Симптоми міопатії можуть превалювати у клінічній картині при системній ССД, ВКВ, ревматоїдному артриті, синдромі Шегрена, системних васкулітах. Загалом, для перехресних синдромів характерні висока частота феномену Рейно та поліартриту, дуже високі титри антинуклеарного фактора (але відсутність міозитспецифічних аутоантитіл), гарна відповідь на глюкокортикоїди. У хворих на поліміозит у поєднанні з ССД у сироватках виявляють AT PM/Scl. Підвищення активності КФК при перехресних міозитних синдромах при запаленнях м'язів виражене так само, як і при ідіопатичному поліміозиті/дерматоміозіті, а гістологічні зміни в м'язах подібні до тих, що спостерігаються при ідіопатичних формах захворювання.
Ознаки міозиту м'язів
Ознаки міозиту при злоякісних новоутвореннях при запаленні м'язів
Симптоми міозиту, асоційовані з пухлинами (пухлинний, або паранеопластичний, міозит), становлять приблизно 20% від усіх випадків запальних міопатій. На фоні злоякісних новоутвореньчастіше розвивається дерматоміозит, ніж поліміозит. Співвідношення кількості хворих чоловіків і кількості хворих жінок становить 1:1. Пухлини можуть розвиватися до появи ознак міопатії, миттєво або після них. Частота злоякісних новоутворень при поліміозит/дерматоміозит в 12 разів вище, ніж у загальній популяції.
З клінічної точки зору розвиток у хворих на васкуліт (або шкірний некроз) або аміотрофічний дерматоміозит збільшує ймовірність пухлинного міозиту, а формування легеневого фіброзу, поява міозитспецифічних аутоантитіл та інших системних проявів знижують її. Локалізація та тип пухлин при запаленні м'язів, що асоціюються з міозитом, збігаються з їх розподілом за частотою у відповідній віковій групі. Тим не менш, пухлинний міозит частіше асоційований з раком яєчника і носоглотки. При підозрі на пухлинний міозит слід провести додаткове діагностичне обстеження, що включає визначення простатспецифічного АГ (у чоловіків), СА-125 (АГ пухлини яєчника).



